Bulk RNA-seq (7): From ENSEMBL ID Cleanup to Gene Name Conversion
Note
In the realm of RNA-seq analysis, one often encounters the challenge of working with ENSEMBL IDs that include decimal points, a result of gene versioning in databases. Furthermore, for meaningful biological interpretation, it’s essential to convert these ENSEMBL IDs to more recognizable gene names. This blog post will guide you through the process of cleaning up these IDs and converting them for enhanced clarity in your analyses.
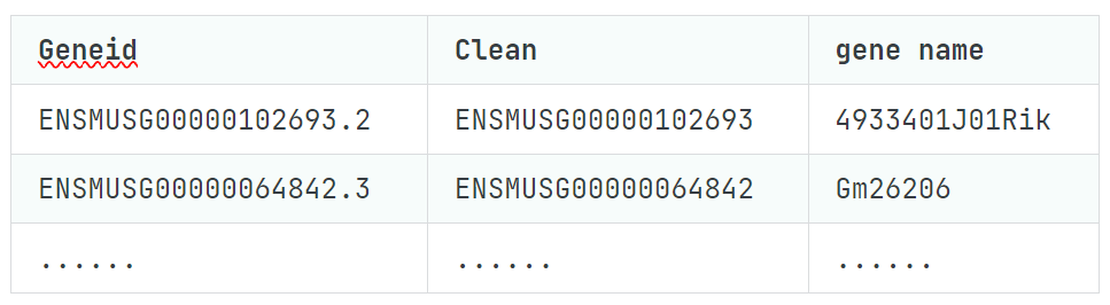
| Geneid | Clean | gene name |
|---|---|---|
| ENSMUSG00000102693.2 | ENSMUSG00000102693 | 4933401J01Rik |
| ENSMUSG00000064842.3 | ENSMUSG00000064842 | Gm26206 |
| …… | …… | …… |
Preparing Your Workspace with Essential R Packages
Before diving into the data manipulation, ensure that your R environment is equipped with the necessary packages. Here’s how to install them:
install.packages("stringr")
if (!requireNamespace("BiocManager", quietly = TRUE))
install.packages("BiocManager")
BiocManager::install("clusterProfiler")
BiocManager::install("biomaRt")
In the RNA-seq analysis, one often encounters the challenge of working with ENSEMBL IDs that include decimal points, a result of gene versioning in databases. Furthermore, for meaningful biological interpretation, it’s essential to convert these ENSEMBL IDs to more recognizable gene names. This blog post will guide you through the process of cleaning up these IDs and converting them for enhanced clarity in your analyses.
Preparing Your Workspace with Essential R Packages
Before diving into the data manipulation, ensure that your R environment is equipped with the necessary packages. Here’s how to install them:
Load the installed packages into your R session:
install.packages("stringr")
if (!requireNamespace("BiocManager", quietly = TRUE))
install.packages("BiocManager")
BiocManager::install("clusterProfiler")
BiocManager::install("biomaRt")
library(stringr)
library(clusterProfiler)
library(biomaRt)
Cleaning Up ENSEMBL IDs
First, we tackle the issue of decimal points in ENSEMBL IDs which are indicative of gene versions. For many downstream analyses, these versions are unnecessary and can be removed for simplicity.
# Load your count data
ENSEM <- read.table(file = "featurecounts.txt", header = TRUE, sep = "\t")
head(ENSEM$Geneid)
# Remove decimal points from ENSEMBL IDs
ENSEM$Geneid = unlist(str_split(ENSEM$Geneid, "[.]", simplify = TRUE))[, 1]
head(ENSEM$Geneid)
# Save the cleaned data
write.csv(ENSEM, file = "totalexonreads.csv", quote = FALSE, row.names = FALSE)
Converting ENSEMBL IDs to Gene Names
With cleaned ENSEMBL IDs, the next step is to convert them into gene names, facilitating easier identification and interpretation of the genes involved in your study.
# Reload the cleaned data
mycounts <- read.csv(file = "totalexonreads.csv", row.names = 1, header = TRUE)
head(mycounts)
# Initialize the ENSEMBL biomaRt dataset for mouse
ensembl <- useMart(biomart = "ensembl", dataset = "mmusculus_gene_ensembl")
# Specify the attributes of interest: ENSEMBL IDs and gene symbols
attributes <- listAttributes(ensembl)
attr <- c("ensembl_gene_id", "mgi_symbol")
# Perform the ID conversion
value <- ENSEM$Geneid
ids <- getBM(attributes = attr, filters = "ensembl_gene_id", value = value, mart = ensembl)
By employing the biomaRt package, you can seamlessly map ENSEMBL IDs to their corresponding gene symbols. This conversion not only aids in the readability of your results but also in their biological interpretation.
This ids will be very important for us to generate volcano plot!