Bulk RNA-seq (4): Mapping and Quality Control with STAR, Qualimap, and featureCounts
Note
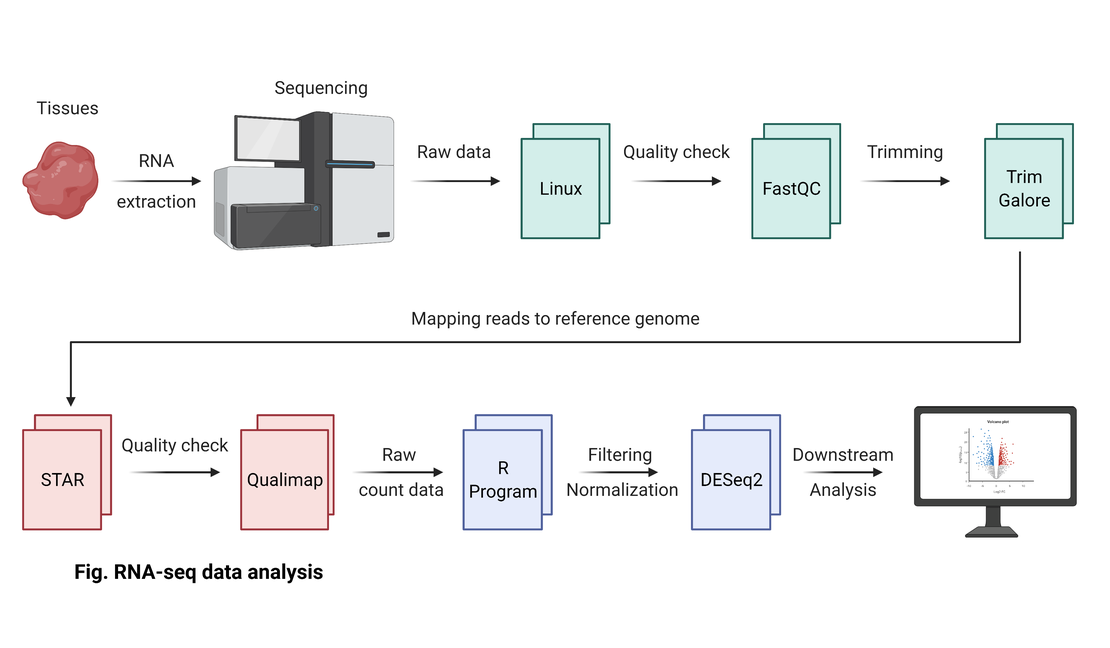
This section of the blog series will guide you through mapping RNA-seq read files to a genome index using STAR, performing quality control (QC) with Qualimap, and generating count data with featureCounts. Each step is crucial for ensuring the accuracy and reliability of RNA-seq data analysis.
1. Installing STAR
STAR (Spliced Transcripts Alignment to a Reference) is a widely used aligner for RNA-seq data. To install STAR, follow the official manual :
# Download the latest STAR source from releases
wget https://github.com/alexdobin/STAR/archive/2.7.9a.tar.gz
tar -xzf 2.7.9a.tar.gz
cd STAR-2.7.9a/source
make STAR
# Verify the installation
STAR
2. Generating Genome Index
Before mapping, it’s essential to generate a genome index. I used mouse genome data from GENCODE for this purpose.


To generate the genome index, create a new folder named index and run STAR:
#create a new folder named as index
mkdir /home/xxx/reference/index
#running STAR for the generation of index
STAR --runThreadN 10
--runMode genomeGenerate
--genomeDir index/
--genomeFastaFiles GRCh38.p13.genome.fa
--sjdbGTFfile gencode.v38.annotation.gtf
--sjdbOverhang 35
3. Mapping with STAR in Two-Pass Mode
Mapping in two-pass mode allows STAR to use splice junctions discovered in the first pass to improve alignment in the second pass:
STAR --runThreadN 10 \
--runMode alignReads \
--readFilesCommand zcat \
--twopassMode Basic \
--outSAMtype BAM SortedByCoordinate \
--genomeDir ~/reference/genome/grcm39/index/ \
--readFilesIn 2_R1_val_1.fq.gz 2_R2_val_2.fq.gz \
--outFileNamePrefix ~/wkdir
Mapping typically takes about 6 minutes per sample. The log.final.out file provides important statistics like mapping rate and number of multi-mappers.

4. Quality Control with Qualimap
Qualimap is a tool that provides additional QC metrics for mapping data. To run Qualimap on your BAM files:
~/qualimap_v2.2.1/qualimap rnaseq -bam ~/dir/xxx.bam -gtf ~/dir/annotation.gtf -outdir ~/dir/WT1 --java-mem-size=8G
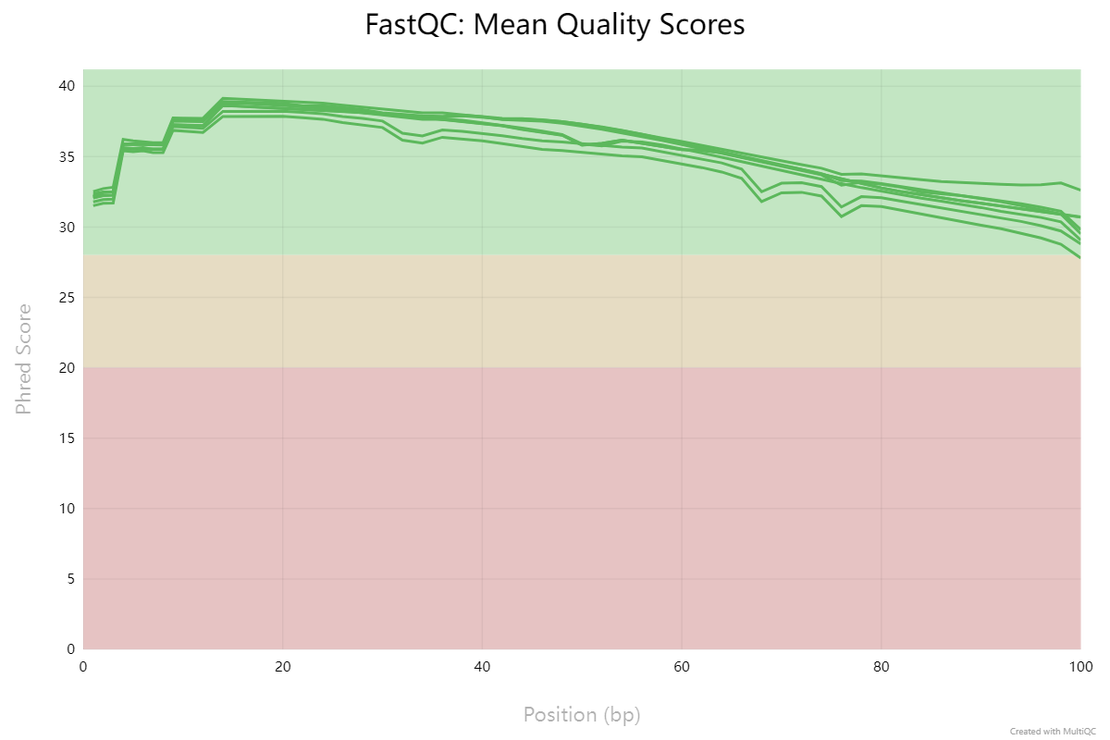
Qualimap reports include the percentage of reads mapped and the proportion of reads mapped to exonic regions.
5. Generating Count Data with featureCounts
featureCounts quantifies read counts per gene based on the mapping results:
featureCounts -s 2 -p -t gene -g gene_id -a ~/dir/annotation.gtf -o counts.txt *.bam
Remove unnecessary columns for a cleaner count table:
cut -f1,7-100 counts.txt > featurecounts.txt
Through these steps, you can map RNA-seq reads, perform detailed quality control, and generate count data for downstream analysis, ensuring a robust foundation for exploring gene expression.