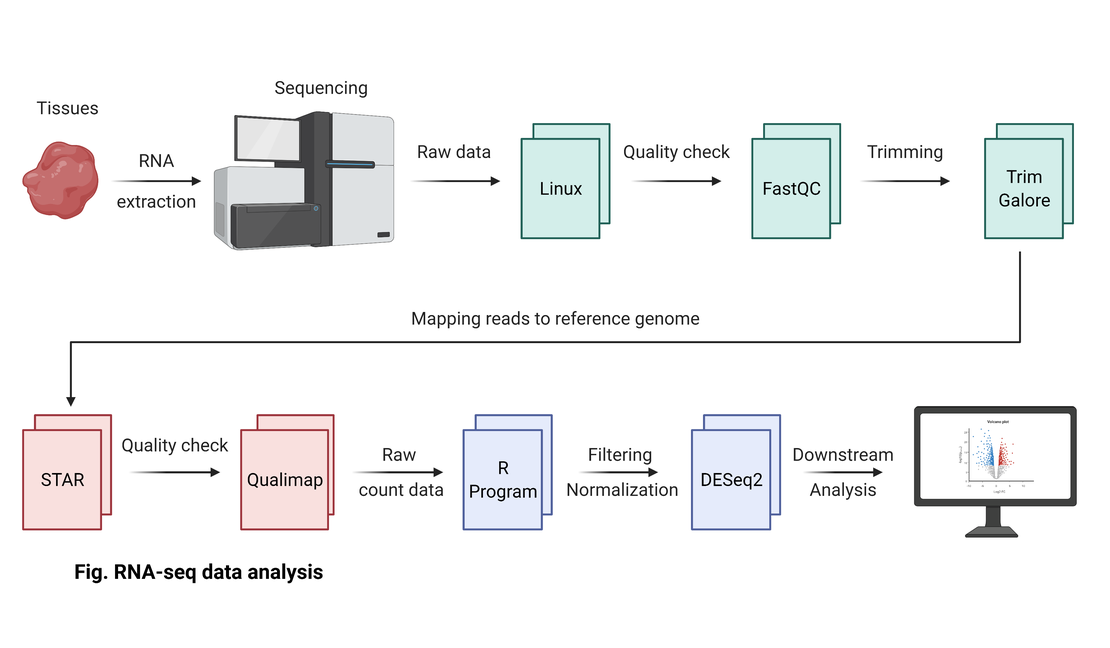
bulk RNA-seq(1):Concatenation of raw read files
Concatenation of Raw Read Files
To begin with, my operating environment is Linux Ubuntu 20.04.4, and I am working with 64 raw read fastq.gz files.
What we need to accomplish first is the concatenation of 4 files into a single file. For instance:
| File name (before) | File name(after) |
|---|---|
| LAB_273_17_…_L001_R1_001.fastq.gz | LAB_273_17_R1.fastq.gz |
| LAB_273_17_…_L002_R1_001.fastq.gz | |
| LAB_273_17_…_L003_R1_001.fastq.gz | |
| LAB_273_17_…_L004_R1_001.fastq.gz |
Tip
Lane Splitting: To increase throughput and reduce experimental bias, a sample’s DNA or RNA library might be sequenced across several lanes. This results in a separate data file for each lane, leading to multiple R1.fastq.gz files for a single sample.
To achieve this in Linux, open the terminal and execute the following script:
for name in *.fastq.gz; do
printf '%s\n' "${name%_*_*_R[12]*}"
done | uniq |
while read prefix; do
cat "$prefix"*R1*.fastq.gz > "${prefix}_R1.fastq.gz"
cat "$prefix"*R2*.fastq.gz > "${prefix}_R2.fastq.gz"
done
Subsequently, verify if the files have been successfully concatenated.