bulk RNA-seq(3):Trim the read files using trim_galore
Tip
We will install Trim Galore and trim the merged read files to improve the quality of our data.
1. The Importance of Trimming
Quality trimming reduces the overall number of reads but increases the total and proportion of uniquely mapped reads, yielding more useful data for downstream analyses. However, overly aggressive trimming can adversely affect subsequent analyses.
:man_teacher: Trimming removes low-quality reads! It’s essential before mapping!
:woman_teacher: Be cautious! Trimming might remove critical reads, including those considered low quality.
:bulb: The decision to trim should be based on your project needs and FastQC results.
2. Assessing Read Quality with FastQC
Refer to my previous article for how to use FastQC.
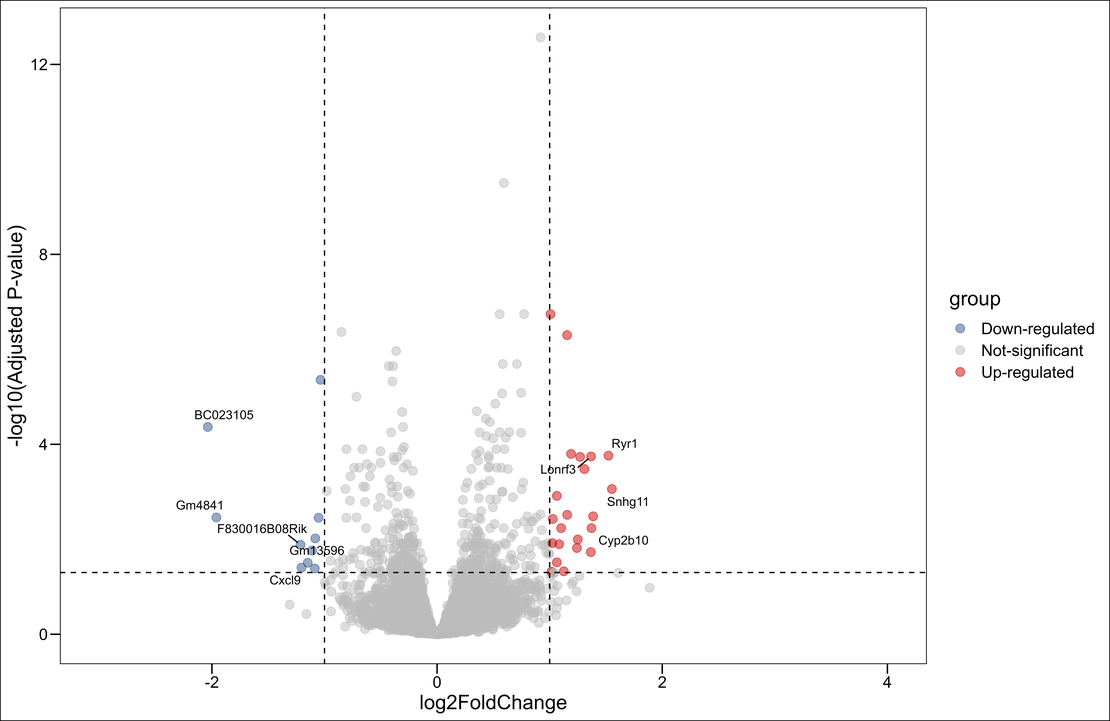
2.1 Per Base Sequence Quality

:warning: The figure above shows that the last 10 base pairs are of low quality, especially the very last one (marked with a red circle). Therefore, I’ve decided to trim this sample.
3. Trimming with Trim Galore
Note
Our guide follows the Trim Galore instructions .
3.1 Installing Trim Galore
Visit the download site on GitHub.
# Download the trim galore tar.gz file
tar -zxvf 0.6.7.tar.gz
Verify the installation:
trim_galore -version
trim_galore --help
3.2 Running Trim Galore
trim_galore -q 25 --phred 33 --stringency 3 --length 30 --paired 1_R1.fastq.gz 1_R2.fastq.gz --gzip -o./cleandata
Trimming a paired sample typically takes about 15 minutes.
3.3 Automating Trimming with a Shell Script
I have created a shell script to automate the trimming process with Trim Galore. This script is designed to trim paired-end sequencing files for a series of samples by specifying their prefixes.
#!/bin/bash
# Specify the output directory where trimmed files will be stored
OUT_DIR=~/OUT_DIR
# List of sample prefixes to process
Prefixes=(
"LAB_410_13"
"LAB_410_14"
"LAB_410_15"
"LAB_410_16"
"LAB_410_17"
"LAB_410_18"
"LAB_410_19"
"LAB_410_20"
"LAB_410_21"
"LAB_410_22"
"LAB_410_23"
"LAB_410_24"
"LAB_410_25"
"LAB_410_26"
"LAB_410_27"
"LAB_410_28"
)
# Loop through each prefix to construct file names for R1 and R2 reads and perform trimming
for PREFIX in "${Prefixes[@]}"
do
R1=${PREFIX}_R1.fastq.gz
R2=${PREFIX}_R2.fastq.gz
# Execute Trim Galore with specified parameters
trim_galore -q 25 --phred33 --stringency 3 --length 30 --paired $R1 $R2 --gzip -o $OUT_DIR
done
Script Explanation and Usage:
- Output Directory (
OUT_DIR): The path where the script saves the trimmed files. Modify this to suit your project structure. - Sample Prefixes (
PrefixesArray): This array holds the identifiers for the sample sets to be processed. These identifiers are crucial for generating the filenames for paired-end reads. - Processing Loop: For each sample identifier in the
Prefixesarray, the script constructs the filenames for R1 and R2 reads and executes Trim Galore to trim these files. - Trim Galore Command: The script runs Trim Galore with parameters set for quality (
-q 25), encoding (--phred33), stringency (--stringency 3), minimum length (--length 30), and output compression (--gzip), directing the output to the specified directory.
To run this script:
- Save the script into a file, e.g.,
trim_script.sh. - Make the script executable by running
chmod +x trim_script.sh. - Execute the script with
./trim_script.shin your terminal.
This script aims to streamline the preprocessing of multiple RNA-seq samples by automating the trimming process, ensuring consistency and saving valuable time during quality control checks.
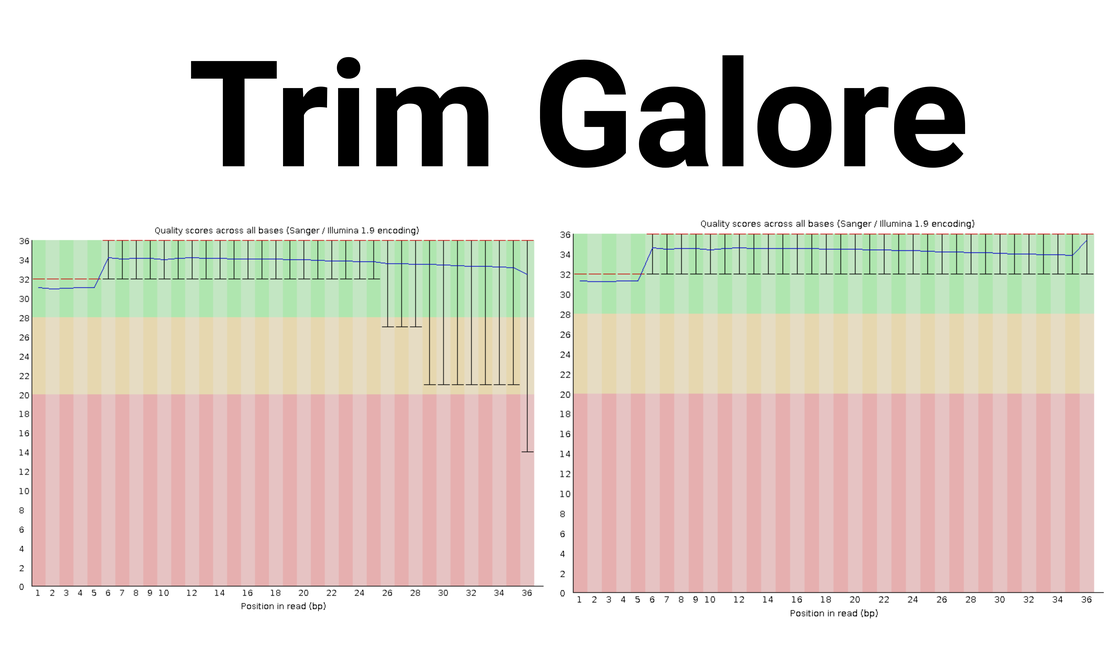
4. Quality Check Post-Trimming with FastQC & MultiQC
It’s crucial to reassess the quality of your reads after trimming.
fastqc *.gz
multiqc .

The quality significantly improves after trimming! :v: